
Exhibit 99.2
ARTICLE IN PRESS
Five-Year Safety and Performance Results from the Argus II Retinal Prosthesis System Clinical Trial
Lyndon da Cruz, MD,1 Jessy D. Dorn, PhD,2 Mark S. Humayun, MD, PhD,3 Gislin Dagnelie, PhD,4 James Handa, MD,4 Pierre-Olivier Barale, MD,5 José-Alain Sahel, MD,5,6,7 Paulo E. Stanga, MD,9 Farhad Hafezi, MD, PhD,3,8,10 Avinoam B. Safran, MD,6,10 Joel Salzmann, MD,10 Arturo Santos, MD, PhD,11 David Birch, PhD,12 Rand Spencer, MD,13 Artur V. Cideciyan, PhD,14 Eugene de Juan, MD,15 Jacque L. Duncan, MD,15 Dean Eliott, MD,3,16 Amani Fawzi, MD,3,17 Lisa C. Olmos de Koo, MD,3 Allen C. Ho, MD,18 Gary Brown, MD,18 Julia Haller, MD,4,18 Carl Regillo, MD,18 Lucian V. Del Priore, MD,19 Aries Arditi, PhD,20 Robert J. Greenberg, MD, PhD,2 for the Argus II Study Group*
Purpose: The Argus II Retinal Prosthesis System (Second Sight Medical Products, Inc, Sylmar, CA) was developed to restore some vision to patients blind as a result of retinitis pigmentosa (RP) or outer retinal degeneration. A clinical trial was initiated in 2006 to study the long-term safety and efficacy of the Argus II System in patients with bare or no light perception resulting from end-stage RP.
Design: Prospective, multicenter, single-arm clinical trial. Within-patient controls included the nonimplanted fellow eye and patients’ native residual vision compared with their vision with the Argus II.
Participants: Thirty participants in 10 centers in the United States and Europe.
Methods: The worse-seeing eye of blind patients was implanted with the Argus II. Patients wore glasses mounted with a small camera and a video processor that converted images into stimulation patterns sent to the electrode array on the retina.
Main Outcome Measures: The primary outcome measures were safety (the number, seriousness, and relatedness of adverse events) and visual function, as measured by 3 computer-based, objective tests. Secondary measures included functional vision performance on objectively scored real-world tasks.
Results: Twenty-four of 30 patients remained implanted with functioning Argus II Systems at 5 years after implantation. Only 1 additional serious adverse event was experienced after the 3-year time point. Patients performed significantly better with the Argus II on than off on all visual function tests and functional vision tasks.
Conclusions: The 5-year results of the Argus II trial support the long-term safety profile and benefit of the Argus II System for patients blind as a result of RP. The Argus II is the first and only retinal implant to have market approval in the European Economic Area, the United States, and Canada. Ophthalmology 2016; n :1–7 © 2016 by the American Academy of Ophthalmology. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
 |
*Supplemental material is available at www.aaojournal.org. |
The last decade has seen a significant number of new retinal treatment paradigms commencing clinical trials. These have included gene therapy,1 stem cell transplantation,2 and electronic neural prostheses in different locations in the eye.3–5 Although all of these approaches hold promise, only retinal prostheses have reached the market for the restoration of some visual function in patients blind as a result of retinitis pigmentosa (RP). The Argus II Retinal Prosthesis System (Second Sight Medical Products, Inc, Sylmar, CA) was the first and remains 1 of only 2 retinal prostheses to be approved for commercialization in the European Economic Area (receiving CE Mark in 2011) and the only prosthesis to date to receive Food and Drug Administration approval for commercialization in the United States (Humanitarian Device Exemption approval in 2013) and to receive Health Canada approval (in 2014).
Thirty patients implanted with the Argus II System for the Argus II feasibility study (clinicaltrials.gov identifier, NCT00407602) are being followed for 10 years in a long-term follow-up clinical study. All enrolled patients now have reached at least 5 years after implantation; this report includes safety and efficacy data for all enrolled patients for that period.
| © 2016 by the American Academy of Ophthalmology | http://dx.doi.org/10.1016/j.ophtha.2016.06.049 |
| This is an open access article under the CC BY-NC-ND license | ISSN 0161-6420/16 |
| (http://creativecommons.org/licenses/by-nc-nd/4.0/). Published by Elsevier Inc. |
ARTICLE IN PRESS
Ophthalmology Volume n, Number n, Month 2016
Methods
The Argus II System
The Argus II Retinal Prosthesis System is a visual prosthesis with implanted and external components (Fig 1). Patients wear a pair of glasses with a small camera mounted in the frame connected via a cable to a video processing unit worn on the belt or on a shoulder strap. Implanted components include a hermetically sealed enclosure for the electronics that, along with a receiving antenna, is secured to the eye with a scleral band and sutures, and an array of 60 electrodes that is inserted into the eye and tacked over the macula. When the system is turned on, the visual information collected by the camera is received, processed, and converted into a brightness map in real time by the video processing unit. Power and data are sent via a radio-frequency telemetry link from an external antenna on the glasses to the receiving antenna on the eye. The brightness values in the video are converted into stimulation current amplitudes on each of the 60 electrodes; activated retinal neurons produce action potentials that travel through the remaining visual system and are perceived as patterns of light by the patients.
Surgical Procedure
The Argus II was implanted in the worse-seeing eye of each patient. The surgical procedure has been reported in detail elsewhere6,7; herein, we provide a summary of the main steps. A 3603 limbal conjunctival peritomy was performed. The receiving coil was inserted under the lateral rectus muscle and extended into the inferotemporal quadrant, whereas the electronics case was placed in the superotemporal quadrant. The scleral band continued under the inferior, medial, and superior rectus muscles. Suture tabs on the implant allowed fixation of the implant to the sclera, and a Watzke sleeve (Labtician Ophthalmics, Inc, Oakville, Canada) and mattress sutures or scleral tunneling secured the scleral band in the nasal quadrants. Core and peripheral vitrectomies were performed, and a temporal sclerotomy of approximately 5 mm was made to allow the introduction of the 60-electrode array into the eye. The array was placed over the macula and tacked to the retina with a custom-made, spring-tension, metallic tack (Second Sight Medical Products, Inc). The trans-scleral passage of the cable was sealed with sutures, and all other sclerotomies were closed. An allograft (processed pericardium; aponeurosis in France) was sutured over the implant to reduce the risk of conjunctival irritation or erosion, and the Tenon’s capsule and conjunctiva were closed.
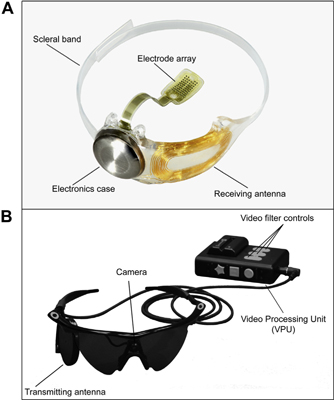
Figure 1. Photographs of the Argus II Retinal Prosthesis System (Second Sight Medical Products, Inc, Sylmar, CA): (A) the implanted components of the system and (B) the external (body-worn) components of the system.
Study Design
The Argus II clinical trial was a prospective, single-arm, non-randomized study. A sample size of 30 was chosen as sufficient for an analysis of safety and efficacy, taking into account the rarity of the disease under study, RP; estimated prevalence is approximately 1 in 4000 people in developed countries. There were no sham surgeries and all patients were implanted with the Argus II.
Inclusion criteria included: bare light perception or worse vision (>2.9 logarithm of the minimum angle of resolution [logMAR]) in both eyes resulting from profound RP (in the United States) or outer retinal degeneration (in Europe); a history of useful form vision; intact and functioning optic nerve; and 50 years of age or older (later in the trial, this criterion was changed to 25 years or older in the United States and Switzerland and 18 years or older in the United Kingdom and France). Exclusion criteria included: diseases or conditions that may have prevented successful implantation (e.g., axial length out of a certain range) or may have prevented the device from working correctly (e.g., damaged optic nerve function). The trial was and continues to be conducted in accordance with the Declaration of Helsinki and the national regulations for medical device clinical trials in the respective countries where the study is being conducted: the United States, the United Kingdom, France, and Switzerland. The study has been approved by the national ministries of health in these countries and the ethics committees or institutional review boards of participating institutions. All patients signed informed consent to participate. The clinical trial is posted on www.clinicaltrials.gov (where full inclusion and exclusion criteria can be found) under trial registration number NCT00407602.
End Points
The trial end points, summarized here, have been described in detail elsewhere.6–9 The primary end point for safety was the rate, type, and severity of adverse events (AEs) that were related to the surgery or the device. All AEs were collected and reported as necessary to the relevant authorities and ethics committees and received detailed review and adjudication by an independent medical safety monitor. Serious adverse events (SAEs) were distinguished as a subset of AEs according to the regulatory definition. In this trial, events adjudicated as serious met the criteria of “necessitated medical or surgical intervention to preclude permanent impairment or damage to a body structure” or “required hospitalization or prolonged hospitalization.” Nonserious AEs required no treatment or only noninvasive treatment. Thus, a single type of event such as hypotony could be classified as either serious or nonserious depending on how or where it was treated.
Information about device reliability, stability, and robustness over time was gathered by tracking the number of device failures. Data on partial or complete explantation of devices also were captured. Follow-up visits after explantation were performed at 1 day, 1 week, 4 weeks, 3 months, 6 months, and 12 months after explantation except as noted below. Visits after explantation included eye examination, retinal photography, ocular coherence tomography, and fluorescein angiography.
ARTICLE IN PRESS
da Cruz et al • Five-Year Argus II Results
The primary end point for efficacy was visual function, as measured by 3 custom-designed objective assessments. Square localization measured the ability to locate and touch a high-contrast white square of light on a black background on a touch screen monitor; direction of motion assessed patients’ ability to determine and indicate the direction of a high-contrast bar that moved across the monitor; and grating visual acuity measured patients’ visual acuity using square-wave gratings of different spatial frequencies presented on a computer monitor. All assessments were performed with the Argus II turned on and off (with patients’ residual vision only—binocularly for square localization and direction of motion and monocularly for grating visual acuity). Masking of patients was not possible because of the visual and auditory cues produced by the Argus II when turned on.
Square localization and direction of motion were analyzed in terms of their mean error (the difference between the stimulus and response, in centimeters and degrees, respectively). For the analysis as a group, results from all patients were pooled at each time point, such that mean error indicated the overall performance of the group. For individual analyses, a 2-tailed t test assuming unequal variances indicated whether the mean error with the system turned on was significantly different from system off for each patient (P < 0.05). Grating visual acuity was measured on a scale of 2.9 to 1.6 logMAR. Patients who performed no better than chance at 2.9 logMAR were scored worse than 2.9 logMAR. The percentage of patients who scored better than 2.9 logMAR was compared for the 2 conditions.
Secondary end points included the door task, a real-world assessment in which patients attempted to walk to and touch a large piece of contrasting felt (simulating a door) on a wall; the line task, in which patients followed a white line painted on black tiles; the mass of activity inventory, a questionnaire designed to measure changes in functional vision; the functional low-vision observer-rated assessment, an assessment performed by trained low-vision rehabilitation specialists; and the vision-related quality of life questionnaire, designed to measure the quality of life of those with vision impairments. The functional low-vision observer-rated assessment and vision-related quality of life questionnaire were performed only through postimplant year 3 (as per the clinical trial protocol), and the functional low-vision observer-rated assessment has been described and reported elsewhere.10,11 The activity inventory was not validated fully in this patient population (e.g., with very low-vision patients), and as such, the data are not included in this report. The door and line tasks were scored by percent success, that is, the mean percent of correct responses (touching the door or ending at the line) for the system on and off was calculated over the group.
Results
Patient Demographics
Enrollment of 30 patients at 10 centers was completed in 26 months (between June 2007 and August 2009), including a pause in enrollment of approximately 6 months after the first 15 patients were implanted. Basic demographics are shown in Table 1.
| Table 1. Demographics of Enrolled Participants | ||
| No. of participants | 30 | |
| Retinitis pigmentosa, no. | 29 (including 1 LCA) | |
| Choroideremia, no. | 1 | |
| BLP, no. | 29 | |
| NLP, no. | 1 | |
| Gender, no. | ||
| Female | 9 | |
| Male | 21 | |
| Mean age ± SD at time of implantation, yrs | 58±10 | |
| Age range at time of implantation, yrs | 28–77 | |
| Years since diagnosis at time of implantation, mean ± SD | 35.23±11.7 | |
| Years of BLP at time of implantation, mean ± SD (n = 15) | 15.9±37.9 |
BLP = bare light perception; LCA = Leber Congenital Amaurosis;
NLP =no light perception; SD = standard deviation.
Patients Lost to Follow-up
No patients were lost to follow-up completely as of 5 years after implantation. However, the number of patients included in the analysis of safety and efficacy did decline over time. Safety data were gathered to 5 years for 27 patients, with drop-out occurring for explanted patients at 1.2 years, 3.5 years, and 4.3 years. Performance data were gathered for 21 or 20 patients at 5 years after implantation as described later.
Safety
Previous reports presented SAE data at 1 year and 3 years after implantation.6,7 Here, we reprint the 0- to 3-year cumulative SAE rates and report SAEs that occurred up to 5 years after implantation (Table 2). As of 5 years after implantation, 60% of patients (18/30) had experienced no device- or surgery-related SAEs. There were 24 SAEs among 12 patients. (See Table S1, available online at www.aaojournal.org, for SAE occurrences per post-implant year.)
All SAEs were treatable with standard ophthalmic approaches, and there were no lost eyes (enucleated) in the study. As shown in Table 2, only 1 additional SAE had occurred up to year 5 since the last analysis at 3 years after implantation. A rhegmatogenous retinal detachment was noted in the implanted eye of 1 patient during a routine follow-up visit approximately 4.5 years after implantation. The detachment remained stable for more than 1 year, when neovascular glaucoma associated with rubeosis was noted. Medication did not decrease the intraocular pressure. Thus, the patient underwent a pars plana vitrectomy, removal of an epiretinal membrane, fluid–air exchange, and injection of silicone oil. Two weeks after surgery, the intraocular pressure returned to normal and the rubeosis and retinal detachment resolved. One patient died at 6 years after implantation of natural causes unrelated to the Argus II.
Device Reliability and Stability
As of 5 years after implantation, 2 Argus II implants had failed, both because of a progressive loss of ability to maintain the radio-frequency link between the external antenna on the glasses and the receiving antenna implanted on the eye. Both devices failed at approximately 4 years after implantation. The failures are believed to be the result of a gradual exposure of a portion of the receiving antenna, possibly because of damage during surgery. The devices remained implanted to continue collecting long-term safety data for the duration of the clinical trial; thus, the root causes cannot be confirmed at this time.
ARTICLE IN PRESS
Ophthalmology Volume n, Number n, Month 2016
Table 2. Serious Adverse Event Rates (Cumulative) to 3 and 5 Years after Implantation
| Year 3 | Year 5 | |||||||
| No. (%) with Serious | No. (%) with Serious | |||||||
| Serious Adverse Event Type | Adverse Event | 95% Confidence Interval | Adverse Event | 95% Confidence Interval | ||||
| Conjunctival erosion | 4 (13.3) | 3.1–30.7 | 4 (13.3) | 3.1–30.7 | ||||
| Hypotony | 4 (13.3) | 3.1–30.7 | 4 (13.3) | 3.1–30.7 | ||||
| Conjunctival dehiscence | 3 (10.0) | 2.1–26.5 | 3 (10.0) | 2.1–26.5 | ||||
| Presumed endophthalmitis | 3 (10.0) | 2.1–26.5 | 3 (10.0) | 2.1–26.5 | ||||
| Retack | 2 (6.7) | 0.8–22.1 | 2 (6.7) | 0.8–22.1 | ||||
| Retinal detachment | ||||||||
| Rhegmatogenous | 1 (3.3) | 0.1–17.2 | 2 (6.7) | 0.8–22.1 | ||||
| Tractional and serous | 1 (3.3) | 0.1–17.2 | 1 (3.3) | 0.1–17.2 | ||||
| Retinal tear | 1 (3.3) | 0.1–17.2 | 1 (3.3) | 0.1–17.2 | ||||
| Uveitis | 1 (3.3) | 0.1–17.2 | 1 (3.3) | 0.1–17.2 | ||||
| Keratitis, infective | 1 (3.3) | 0.1–17.2 | 1 (3.3) | 0.1–17.2 | ||||
| Corneal melt | 1 (3.3) | 0.1–17.2 | 1 (3.3) | 0.1–17.2 | ||||
| Corneal opacity | 1 (3.3) | 0.1–17.2 | 1 (3.3) | 0.1–17.2 | ||||
| Total | 23 | 24 | ||||||
Device Explantations
There were 3 complete or partial explantations. In 1 patient, as previously reported,7 the implant was removed at 14 months to resolve recurrent conjunctival erosion. Two additional patients requested that their devices be explanted at 3.5 and 4.3 years after implantation. One of these patients had experienced 2 conjunctival erosions that were treated by resuturing the device and closing the conjunctiva. A third instance of conjunctival erosion occurred and the patient chose explantation rather than undergoing a third revision surgery. The entire implant was removed with no serious adverse sequelae. This patient completed follow-up after explantation through 3 months and withdrew study consent at that point. The other patient experienced chronic hypotony and ptosis in the implanted eye and chose explantation for aesthetic reasons and to avoid additional revision surgeries. During the explantation procedure, the cable was cut mid vitreous, the sclerotomy was sutured completely closed, and the extraocular portion of the device and the proximal portion of the cable were removed. The array was left tacked to the retina. No AEs occurred after explantation.
Visual Function
Mean results over time for visual function tasks are shown in Figure 2. The number of patients included in the analysis for each time point is indicated in the axis label. Square localization and direction of motion were introduced partway through the study, so baseline and year 1 follow-up results do not represent a complete data set. Later time points also include fewer patients because of the explantations and device failures described above, as well as a few instances of missed protocol visits in years 4 and 5. Missed visits were the result of health reasons (n = 1), method deviation (n = 1), and consent to safety follow-up only after year 3 or 4 (n = 2). One additional patient did not complete the direction of motion, line task, or door task at year 5 because of fatigue.
As a group, patients perform better on the square localization test with the system on (lower mean error) than when using their residual vision at all time points (Fig 2A). Direction of motion, a more challenging assessment, also showed overall improvement (lower mean error) with the system on at all time points (Fig 2B). Grating visual acuity, the most difficult assessment, also revealed better performance with the system on; with the system off, all results were worse than 2.9 logMAR at yearly time points. With the system on, 27% to 48% of patients scored 2.9 logMAR or better (Fig 2C), depending on the time point.
The patients’ improvements when using the system compared with their residual vision also can be seen on an individual basis, in terms of the percentage of patients who performed significantly better with the system on than off on each assessment. Results at 1 year and 3 years were reported previously; here, the year 3 and year 5 results are compared (Table 3).
Functional Vision
The mean percent success for the orientation and mobility assessments is shown in Figure 3. Performance on the door task was better with the system on than off at all time points (Fig 3A). Similarly, patients’ ability to follow a white line on the floor was much improved when using the system compared with using only their residual vision (Fig 3B).
Discussion
The Argus II was granted regulatory approval in the Euro-pean Economic Area in 2011 and in the United States in 2013 on the basis of earlier results from this clinical trial. However, the original study patients will continue to be followed up out to 10 years to collect very long-term data on the safety and efficacy of this chronically implanted device. Long-term data are ever more important given that the device is becoming available to increasingly large numbers of patients with RP and similar disorders worldwide.
The data from the original group of 30 patients—15 of whom received an earlier design of the device before minor improvements were made7—dcontinue to show clear reliability, safety, and long-term efficacy out to 5 years after implantation. Twenty-four devices remain implanted and functioning. The device stability remains good with only 2 device failures, both of which remain safely implanted but nonfunctional, and 3 explanted devices from among 30 implanted patients. Of the 3 explantations, 1 was carried out to resolve recurrent conjunctival erosion and chronic hypotony. The other 2 explantations were elected by the patients. Although elective, these 2 explantations were prompted by a cascade of SAEs in each patient that have been documented previously.7 In these cases, the patients chose explantation rather than further revision surgeries to address recurrent SAEs. One patient died during the trial.
ARTICLE IN PRESS
da Cruz et al • Five - Year Argus II Results
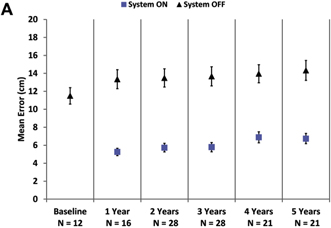
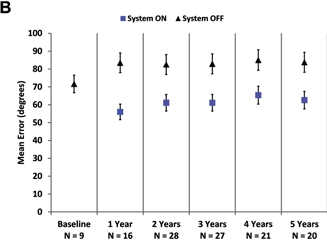
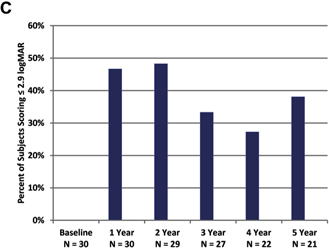
Figure 2. Graphs showing results for (A) square localization, (B) direction of motion, and (C) grating visual acuity at yearly time points. A, B, Mean error with the system on is shown as blue squares; mean error with the system off (with residual vision only) is shown as black diamonds. Error bars indicate standard error. C, The percent of patients scoring 2.9 logarithm of the minimum angle of resolution (logMAR) or better on grating visual acuity with the system on (in the implanted eye) are shown at each time point. There were no patients who scored 2.9 logMAR or better with the system off in the implanted eye.
| Table 3. Individual Visual Function Assessment Results | ||||||||||||||||
| Year 3 | Year 5 | |||||||||||||||
| Visual Function | Significantly Better | Significantly Better | ||||||||||||||
| Assessment | No. | On Than Off (%) | No. | On Than Off (%) | ||||||||||||
| Square localization | 28 | 89.3 | 21 | 80.9 | ||||||||||||
| Direction of motion | 27 | 55.6 | 20 | 50 | ||||||||||||
| Grating visual acuity | 27 | 33.3 | 21 | 38.1 | ||||||||||||
Only 1 new SAE developed between 3 and 5 years after implantation, a rhegmatogenous retinal detachment that was treated successfully and resolved. There were no lost eyes and there was no damaged residual vision in the study. However, it is clear that any chronic implant in the eye carries a continual risk of SAEs. Although outside the scope of this article, additional instances of SAEs were found in 4 patients after the 5-year time point. In 2 patients, these represented recurrences or worsening of previous SAEs; in 2 patients, they were new events (conjunctival erosion and subsequent endophthalmitis in 1 patient, and a rhegmatogenous retinal detachment in another patient). These will be reported in a future article when the dataset is complete and events have been adjudicated. Therefore, it is critical for any patient considering being implanted with the Argus II to understand the long-term and ongoing risks; both the patient and his or her ophthalmologist must commit to at least a yearly evaluation of eye health for as long as the Argus II remains implanted.
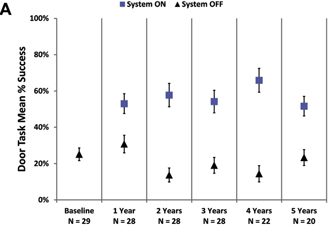
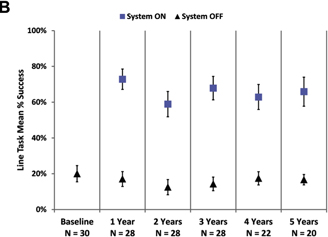
Figure 3. Graphs showing the mean percent success on (A) the door task and (B) the line task with the system on (blue squares) and off (residual vision only, black triangles). Error bars indicate standard error of the mean.
ARTICLE IN PRESS
Ophthalmology Volume -, Number -, Month 2016
Results on a battery of visual function and functional vision assessments indicated continued efficacy of the Argus II out to 5 years after implantation. Patients are still able to locate objects, determine the direction of motion of a moving bar, and perform an acuity task better with the system on than when using only residual vision. The 5-year visual function results are similar to those seen at 3 years, particularly when considering the individual analysis data (e.g., 33% of patients performed grating acuity better with the system on than off at 3 years, and 38% did so at 5 years). Functional vision performance likewise showed sustained improvement with the system on out to 5 years after implantation. It should be noted that 9 to 10 patients did not participate in efficacy testing during the 5-year follow-up period as discussed previously. The resulting smaller numbers may have led to bias in the results at later time points. This potential bias will be evaluated in future reports, such as those on the postapproval studies currently in progress.
In conclusion, as of October 15, 2015, more than 200 patient-years of data had been collected on the 30 patients implanted with the Argus II Retinal Prosthesis System. The longest implant duration to date is 8.4 years, and this device as well as 23 others continue to function, reliably enhancing basic visual function for these patients who otherwise can see almost nothing. The outcome of the functional tests described in this report and the acceptable safety profile of the Argus II in this clinical trial led to its regulatory approval in the European Union, the United States, and Canada. The device has gone on to be implanted in many patients; in many countries, it remains the only currently available treatment for profound vision loss resulting from RP and outer retinal dystrophy. These new long-term data from the original study continue to demonstrate that this therapy remains an option for patients with RP and may allow for stable and reliable restoration of some basic visual function.
Acknowledgments. The authors thank Suber Huang, MD, who served as the independent medical safety monitor for the Argus II Clinical Trial.
References
| 1. | Petrs-Silva H, Linden R. Advances in gene therapy technologies to treat retinitis pigmentosa. Clin Ophthalmol 2014;8: 127–36. |
| 2. | Al-Shamekh S, Goldberg JL. Retinal repair with induced pluripotent stem cells. Transl Res 2014;163:377–86. |
| 3. | Stingl K, Bartz-Schmidt KU, Besch D, et al. Subretinal Visual Implant Alpha IMS–Clinical trial interim report. Vision Res 2015;111(Pt B):149–60. |
| 4. | Kitiratschky VB, Stingl K, Wilhelm B, et al. Safety evaluation of “retina implant alpha IMS”–da prospective clinical trial. Graefes Arch Clin Exp Ophthalmol 2015;253: 381–7. |
| 5. | Ayton LN, Blamey PJ, Guymer RH, et al. First-in-human trial of a novel suprachoroidal retinal prosthesis. PLoS One 2014;9: 1 –26. e115239. |
| 6. | Ho AC, Humayun MS, Dorn JD, et al. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology 2015;122:1547–54. |
| 7. | Humayun MS, Dorn JD, da Cruz L, et al. Interim results from the international trial of Second Sight’s visual prosthesis. Ophthalmology 2012;119:779–88. |
| 8. | Ahuja AK, Dorn JD, Caspi A, et al. Blind subjects implanted with the Argus II retinal prosthesis are able to improve performance in a spatial-motor task. Br J Ophthalmol 2011;95:539–43. |
| 9. | Dorn JD, Ahuja AK, Caspi A, et al. The detection of motion by blind subjects with the epiretinal 60-electrode (Argus II) retinal prosthesis. JAMA Ophthalmol 2013;131:183–9. |
| 10. | Geruschat DR, Flax M, Tanna N, et al. FLORA: phase I development of a functional vision assessment for prosthetic vision users. Clin Exp Optom 2015;98:342–7. |
| 11. | Geruschat DR, Richards TP, Arditi A, et al. An analysis of observer-rated functional vision in patients implanted with the Argus II Retinal Prosthesis System at three years. Clin Exp Optom 2016;99:227–32. |
Footnotes and Financial Disclosures
| Originally received: April 5, 2016. | |
| Final revision: June 4, 2016. | |
| Accepted: June 17, 2016. | |
| Available online: nnn. | Manuscript no. 2016-691. |
1 Department of Vitreoretinal Surgery, Moorfields Eye Hospital, NHS Foundation Trust, NIHR Moor3elds Biomedical Research Centre, London, United Kingdom and Department of Brain Science, University College London (UCL).
2 Second Sight Medical Products, Inc, Sylmar, California.
3 University of Southern California, Los Angeles, California.
4 Wilmer Eye Institute, Johns Hopkins University, Baltimore, Maryland.
5 Centre Hospitalier National d’Ophtalmologie des Quinze-Vingts, Paris, France.
6 Sorbonne Universities–UPMC Paris-6, and Institut de la Vision, Paris, France.
7 Rothschild Ophthalmology Foundation, Paris, France.
8 ELZA Institute, Zurich, Switzerland.
9 Manchester Royal Eye Hospital, Manchester Vision Regeneration (MVR) Laboratory at NIHR/Wellcome Trust Manchester CRF, and Manchester Academic Health Science Centre and Centre for Ophthalmology and Vision Research, Institute of Human Development, University of Manchester, Manchester, United Kingdom.
10 Hôpitaux Universitaires de Genève, Geneva, Switzerland.
11 Centro de Retina Medica y Quirúrgica, SC, and Tecnologico de Mon-terrey, Guadalajara, Mexico.
12 Retina Foundation of the Southwest, Dallas, Texas.
13 Texas Retina Associates, Dallas, Texas.
ARTICLE IN PRESS
da Cruz et al 3 Five-Year Argus II Results
14 Scheie Eye Institute, University of Pennsylvania, Philadelphia, Pennsylvania.
| 15 | University of California, San Francisco, San Francisco, California. |
16 Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts.
17 Feinberg School of Medicine, Northwestern University, Chicago, Illinois.
| 18 | Wills Eye Hospital, Philadelphia, Pennsylvania. |
19 Columbia University, New York, and Storm Eye Institute, Charleston, South Carolina.
20 Lighthouse Guild, New York, and Visibility Metrics, LLC, Chappaqua, New York.
*Group members and affiliations are listed in the Supplemental Appendix (available at www.aaojournal.org).
Financial Disclosure(s):
The author(s) have made the following disclosure(s): L.C: Consulting – Second Sight Medical Products, Inc., (Sylmar, CA).
J.D.D.: Employee and financial interest – Second Sight Medical Products, Inc., Sylmar, CA; Patent e nos. 7,957,811, 8,428,739, 8,554,327, and 5 additional pending.
M.S.H.: Other – Alcon Laboratories, Inc. (Fort Worth, TX); Bausch & Lomb Surgical (Bridgewater, NJ); Clearside (Alpharetta, GA); IRIDEX (Mountain View, CA); Envisia (Research Triangle Park, NC); Reflow (San Clemente, CA); Regenerative Patch Technologies (Glendale, CA); Replenish (Pasadena, CA); Second Sight; ICo, Inc. (Akron, OH); Viomedix (Pasadena, CA); Patents – 40 issued and 12 pending.
G.D.: Financial support – Second Sight Medical Products, Inc., (Sylmar, CA); QLT, Inc.
J.H.: Financial support – Second Sight Medical Products, Inc., (Sylmar, CA). P-O.B.: Institution receives funds for clinical trial e Second Sight Medical Products, Inc., (Sylmar, CA).
J-A.S.: Consultant – Pixium Vision (Paris, France); GenSight Biologics (Paris, France); Sanofi-fovea (Paris, France); Genesignal (Lausanne, Switzerland); Vision Medicines (Boston, MA); Equity owner – Pixium Vision; GenSight Biologics; Patent – no. 20130157229 A1 (pending). P.E.S.: Institution receives funds for clinical trial – Second Sight Medical Products, Inc., (Sylmar, CA).
F.H.: Institution receives funds for clinical trial – Second Sight Medical Products, Inc., (Sylmar, CA).
A.B.S.: Institution receives funds for clinical trial – Second Sight Medical Products, Inc., (Sylmar, CA).
D.B.: Institution receives funds for clinical trial and contract – Second Sight Medical Products, Inc., (Sylmar, CA); Contract and scientific advisor StemCell, Inc. (Newark, CA); Financial support – AGTC (Alachua, FL); Shire (Lexington, MA); Acucela (Seattle, WA).
A.V.C.: Financial support – Second Sight Medical Products, Inc., (Sylmar, CA). E.J.: Institution receives funds for clinical trial and stockholder – Second Sight Medical Products, Inc., (Sylmar, CA); Founder and Vice-Chairman ForSight Labs (Menlo Park, CA); Patents – Second Sight Medical Products, Inc.
D.E.: Financial support – Second Sight Medical Products, Inc., (Sylmar, CA).
L.C.O.K.: Scientific advisory board – ScienceBased Health (Houston, TX); Alcon (Fribourg, Switzerland) A.C.H.: Consultant and Financial support – Second Sight Medical Products, Inc., (Sylmar, CA).
G.B.: Institution receives funds for clinical trial – Second Sight Medical Products, Inc., (Sylmar, CA).
J.H.: Institution receives funds for clinical trial – Second Sight Medical Products, Inc., (Sylmar, CA); Financial support – Regenera (Rehovot, Israel); LPath (San Diego, CA); Kalvista (Porter Down, UK); Merck (Kenilworth, NJ); ThromboGenics (Leuven, Belgium); Second Sight Medical Products, Inc.; Advanced Cell Technology (Northbrook, IL); Genetech (South San Francisco, CA).
C.R.: Financial support – Second Sight Medical Products, Inc., (Sylmar, CA).
A.A.: Financial support – Second Sight Medical Products, Inc., (Sylmar, CA).
R.J.G.: Employee and financial interest e Second Sight Medical Products, Inc., Sylmar, CA; Patents – 199 issued and 78 pending.
Supported by the National Institutes of Health, Bethesda, Maryland (grant nos.: 5R01EY012893 [R.J.G., principal investigator], 07, and 1RC3EY020778 [R.J.G., principal investigator]). The clinical trial was sponsored by Second Sight Medical Products, Inc., Sylmar, California. The sponsor participated in the design and conduct of the study; data management, analysis, and interpretation; and preparation and review of the manuscript.
Author Contributions:
Conception and design: da Cruz, Dorn, Humayun, Dagnelie, Handa, Barale, Sahel, Stanga, Hafezi, Safran, Salzmann, Santos, Birch, Spencer, Cide-ciyan, de Juan, Duncan, Eliott, Fawzi, Olmos de Koo, Ho, Brown, Haller, Regillo, Del Priore, Arditi, Greenberg
Analysis and interpretation: da Cruz, Dorn, Humayun, Dagnelie, Handa, Barale, Sahel, Stanga, Hafezi, Safran, Salzmann, Santos, Birch, Spencer, Cideciyan, de Juan, Duncan, Eliott, Fawzi, Olmos de Koo, Ho, Brown, Haller, Regillo, Del Priore, Arditi, Greenberg
Data collection: da Cruz, Dorn, Humayun, Dagnelie, Handa, Barale, Sahel, Stanga, Hafezi, Safran, Salzmann, Santos, Birch, Spencer, Cideciyan, de Juan, Duncan, Eliott, Fawzi, Olmos de Koo, Ho, Brown, Haller, Regillo, Del Priore, Arditi, Greenberg
Obtained funding: none
Overall responsibility: da Cruz, Dorn, Humayun, Dagnelie, Handa, Barale, Sahel, Stanga, Hafezi, Safran, Salzmann, Santos, Birch, Spencer, Cideciyan, de Juan, Duncan, Eliott, Fawzi, Olmos de Koo, Ho, Brown, Haller, Regillo, Del Priore, Arditi, Greenberg
Abbreviations and Acronyms:
AE = adverse event; LCA = Leber Congenital Amaurosis; logMAR = logarithm of the minimum angle of resolution; RP = retinitis pigmentosa; SAE = serious adverse event.
Correspondence:
Jessy D. Dorn, PhD, Clinical and Scientific Research, Second Sight Medical Products, 12744 San Fernando Road, Sylmar, CA 91342. E-mail: jdorn@secondsight.com.